
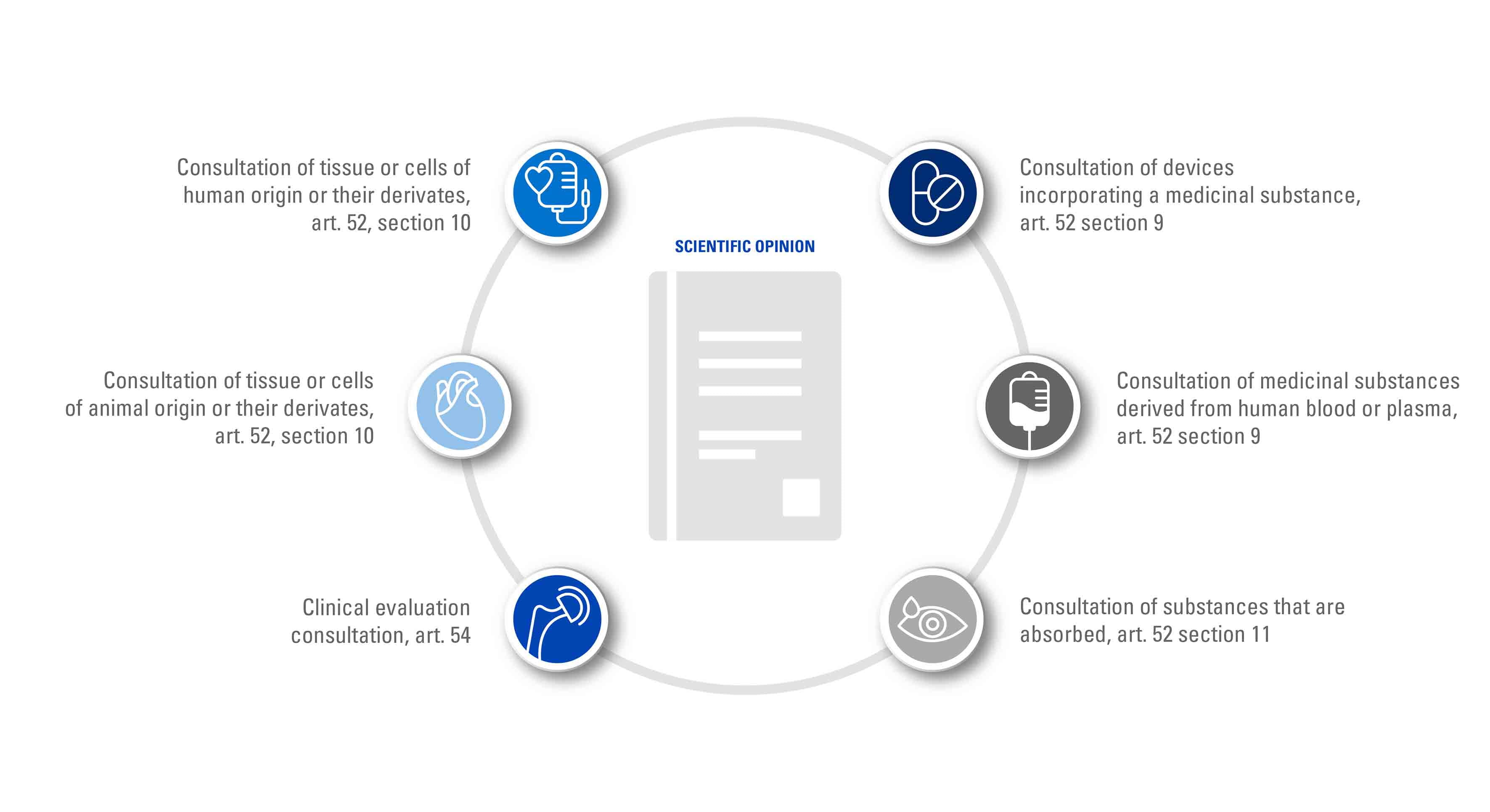
针对某些产品类别,合规评审与审批流程中要求公告机构在颁发欧盟CE证书前,增加与主管机构的确认,以听取专家组意见。MDR法规第52章和第54章中明确规定了专家评审流程。按照器械属性,将由指定主管机构、欧洲药物管理局或欧盟委员会的专家小组参与执行一个或多个评审程序。
当今许多最具创新性的医疗器械实际上是药械结合产品,即器械和药物相结合、药物作用为辅的医疗器械。药械组合产品通常指含药物涂层或预装药品的医疗器械,如带抗菌涂层的导管、带药涂层的支架。其他组合产品,有如含抗生素涂层的球囊导管和骨科水泥、含杀精剂的避孕套等。
按照欧盟医疗器械法规(MDR)附录IX第5.2节的药械结合产品要求,即当器械与药物结合作为整体,且以医疗器械作用为主时,TÜV南德意志集团在颁发欧盟CE技术文档评审证书前,将征求主管机构或欧洲药物管理局的专家组意见。
该程序适用于按照MDR认证的器械,无论它们是否完成MDD或AIMDD下的专家评审程序,或医疗器械是否有变更。
对于MDR下的遗留器械的首次专家评审流程,公告机构应参考专用指南1或根据主管机构要求,向药监机构提交制造商的全套技术文档。因此,制造商应在提交的文件资料时,附上一份完善的变更清单,如以下内容:

如若药用物质无变更,TÜV南德意志集团将提供一份声明,说明自上次专家评审后无新增变更。
针对首次专家评审,主管机构需要在210天内提供评估结果2。对于已在MDD/AIMDD下进行了专家评审程序,并在MDR下首次提交的产品,鉴于上次评审结果的有效性及产品的变更内容,主管机构将考虑审核的深度。
如上所述,专家评审适用于遗留器械,基于制造商判定药物成分不可能作用于人体,则不需要按照MDD/AIMDD进行专家评审。
1 出自欧洲药品管理局就公告机构关于含辅助药物或辅助人血衍生物的医疗器械或有源植入医疗器械,对咨询专家评审流程及文件要求的建议
2 评审周期将不因发补问题延长
欧盟MDR 2017/745法规要求公告机构应就药械结合医疗器械中,对器械具有辅助性(即支持性)作用的药物的质量和安全性,向主管机构征求意见。TüV南德意志集团应做出发证决议前,考虑主管机构的专家组意见。
针对器械中含人血、血浆衍生物或药物,即属于 (EC) 726/2004法规范畴下的药用产品,公告机构应征求欧洲药物管理局的意见。欧洲药物管理局将根据经由TÜV南德意志集团确认的,药械组合产品的生产流程和有效性数据,提供专家组评审意见。
在制造商提交的资料后,TüV南德意志集团将检查文件是否符合EMA/CHMP/578661/2010 rev.1中规定的基本要求。
按照该指南,TÜV南德意志集团将根据产品的预期用途,向欧洲药物管理局递交一份有关辅助性人血衍生物有效性的评审报告。TüV南德意志集团将结合临床评审员的评审以确认产品的有效性。
使用已知的药用物质或已知来源的人血衍生物,且人用医药产品委员会认为不需扩展评估。
当申请加速评审流程时,申请人应至少在首次评审前两至三个月,提前提供支持性文件。申请表格可在欧洲药品管理局网站获取。
经过评审后,人用医药产品委员会/欧洲药物管理局将对该物质的质量和安全性提供专家意见,意见将包括药械结合的临床收益/风险状况。人用医药产品委员会的评审意见将发送至TÜV南德意志集团及其所在欧盟成员国的医疗器械主管当局。
按照欧盟文件EudraLex Notice to Applicants 第2A卷第4章,时间计划参考:在预计提交文件的6个月前,针对TÜV南德意志集团的首次认证项目,可与欧洲药物管理局开展预提交沟通会议。
关于文件提交:
对于使用非活性或无呈现活性的人源组织、细胞的衍生物制造的医疗器械,基于MDR法规附录VIII规则18,适用以下要求:
在申请环节,需确认产品的分类、是否需要欧洲主管当局专家评审流程。制造商必须阐明申请MDR认证的医疗器械是否含有人源性组织或细胞衍生物,是否需要进行专家评审。
在颁发欧盟CE技术文档评估证书前,TüV南德意志集团将根据2004/23/EC指令,就与捐赠、采购和检测无活性或无呈现活性的人源性组织、细胞或其衍生物有关的内容,向成员国指定的主管机构之一(人体组织和细胞主管机构),征求专家组意见。
专家评审流程的期限为120天,如是与捐赠、检测或采购的有关变更,则为60天。
临床评估专家评审流程适用于III类植入器械和IIb类药物输送有源器械(规则12)。
该流程不适用的情况如下:
专家小组应在21天内决定是否提供专家组意见。决策标准如下:
主管机构应在60天内提出专家组意见。
TÜV南德意志集团将合理参考专家小组的评审意见。
如主管机构60天内无意见反馈,TÜV南德意志集团将继续推进器械的发证流程。
当涉及含可吸收物质的器械时,是否需要进行专家评审是一个值得探讨的问题。
无论任何来源(人类、动物、植物或化学品3)的物质,在MDR第一稿公布之前,从不曾是医疗器械界的焦点。每当遇到 “可吸收”的情况,总会听到要求增加专家评审的声音。然而,无论是具有生物效应的物质,或全部、主要被吸收的4,或其他在体内发生化学变化的物质等的器械,都常被误认为需要进行专家评审。其实当中绝大多数物质及其代谢产物,都不一定会被系统地吸收,才能完成预期用途。
如果产品属于MDS 10085的范畴,且为完成预期用途将被系统地吸收,则满足可吸收器械专家评审的前提条件。TÜV南德意志集团随时准备执行主管机构批准的专家评审流程。我们的专业评审员将开展全球范围内的沟通交流,按照与ADME-Tox和相互作用有关的产品数据编制提交文件。换言之,所有关于物质的吸收、分布、代谢、排泄、局部耐受性、毒性、与其他器械、药品或其他物质的相互作用,以及潜在的不良反应特征等内容都应基于质量和安全要求进行评审。我们将向医药产品主管机构(CA)6或欧洲药物管理局(EMA)征求专家组意见,评审反馈预计在收到全部必要文件后150天内提出7。在最终决定中,我们将充分考虑专家组意见,并将我们的发证决定告知主管当局。
当含可吸收物质的器械将作用于人体全身,或至少影响多个器官系统,则有必要进行专家评审。相反,当含可吸收物质的器械虽然属于MDS 1008,但只影响局部组织,则无需进行专家评审。然而,我们有义务评估制造商起草的ADME-Tox和相互作用调查研究。这些评审结果将成为最终技术报告的部分,并影响CE证书的发证决定。
在系统性可吸收物质的专家评审和无无需专家评审的两种情况下,在首次欧盟MDR发证中,都重点关注MDR下可吸收物质的整体质量和安全性。ADME-Tox和相互作用的数据,必须首先满足质量和安全要求。另外,在发生重大变更的情况下8,上述特性应该保持或不受到负面影响。
选择我们为您提供权威的产品认证,将您的时间资源投入于我们资深的专家团队合作中。我们将提供兼备专业能力和灵活度的服务,在高效处理常见的可吸收医疗器械认证的同时,有目的地规划评审时间,以获得欧盟认证。在预申请阶段,请尽早与我们联系,以检查产品的分类、分类规则8和/或规则21的适用性。作为公告机构,我们将为您确认MDN/MDA和MDS代码的适用性,最重要的是,我们将客观地评估可吸收物质专家评审的必要性。您的可吸收物质CE产品证书指日可待!
3 第2001/83/EC号指令,第一篇规定,第1章,第3节
4 医疗器械指南文件MEDDEV 2.4/1注释5和6
5 按照委员会实施细则(EU) 2017/2185,对特定特征的分类编码
6 根据第2001/83/EC号指令
7 MDR附录IX.II.5.4
8 例如:侵入性水平的变更;侵入器械的潜在毒性变更;器械在人体中或对人体作用的部位或介入部位的变更;物质或其代谢产物的吸收变更。
Site Selector
Global
Americas
Asia
Europe
Middle East and Africa

