
17 June 2020
The Central Authority of the German Länder for Health Protection with regard to Medicinal Products and Medical Devices (Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten, ZLG) has now designated TÜV SÜD Product Service as a Notified Body under the new In Vitro Diagnostics Regulation (IVDR). The IVDR regulates market access and monitoring of in vitro diagnostic medical devices and their accessories placed on the EU market. The new regulation came into force in 2017. Products which are already approved must be recertified by 26 May 2022.
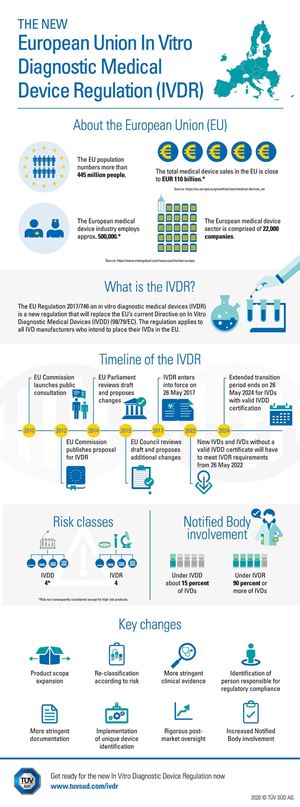 “We are excited about our designation as Notified Body”, says Dr. Andreas Stange, Vice President Medical & Health Services, TÜV SÜD. “In recent months we have expanded our team throughout the world, to deliver comprehensive IVDR activities.” IVDR introduction increases the share of in-vitro diagnostic devices that will have to undergo assessment by a Notified Body in the future from 15 to 90 per cent. Dr. Andreas Stange: “Unless already done, manufacturers of in-vitro diagnostics should get ready for the IVDR immediately to avoid bottlenecks in the testing and certification of their devices.”
“We are excited about our designation as Notified Body”, says Dr. Andreas Stange, Vice President Medical & Health Services, TÜV SÜD. “In recent months we have expanded our team throughout the world, to deliver comprehensive IVDR activities.” IVDR introduction increases the share of in-vitro diagnostic devices that will have to undergo assessment by a Notified Body in the future from 15 to 90 per cent. Dr. Andreas Stange: “Unless already done, manufacturers of in-vitro diagnostics should get ready for the IVDR immediately to avoid bottlenecks in the testing and certification of their devices.”
The new EU Regulation, which replaces the previous IVD Directive, brings massive changes for manufacturers. In-vitro diagnostics are medical devices that enable specimens derived from the human body (such as blood, tissues or saliva) to undergo controlled examination in an artificial environment. In the future, the extended scope of the IVDR will also comprise high-risk devices manufactured and used within a single health institution as well as genetic tests and tests providing information on patients’ genetic disposition.
The IVDR – what is new?
International leading and long-standing experience
TÜV SÜD Product Service is one of the few Notified Bodies designated for both the new IVDR and the previous In-vitro Diagnostics Directive (IVDD). The international service provider is the world’s largest Notified Body for medical devices and in-vitro diagnostic medical devices to be sold in the European Economic Area (EEA). Over authorized 80 experts support manufacturers and marketers, adding safety, quality and sustainability and ensuring the successful market launch of in-vitro diagnostic devices. TÜV SÜD Product Service looks back on a track record of over 30 years in the testing, certification and approval of medical devices and is familiar with the different local regulatory requirements on site. The organisation also has accredited testing laboratories throughout the world.
Further TÜV SÜD information on the IVDR can be found at https://www.tuvsud.com/en/industries/healthcare-and-medical-devices/medical-devices-and-ivd/medical-device-market-approval-and-certification/eu-in-vitro-diagnostic-medical-device-regulation.
Press-contact: Dirk Moser-Delarami
Site Selector
Global
Americas
Asia
Europe
Middle East and Africa


{kind=link}