
TÜV SÜD Product Service is among the world's first certification bodies to receive designation as a Notified Body for the new Medical Device Regulation (MDR) by the Central Authority of the Länder for Health Protection with regard to Medicinal Products and Medical Devices (ZLG). The European Parliament implemented the MDR to improve the EU approval system for medical devices. The regulation which came into force in May 2017, will end its transition period by late May 2020.
 Stricter requirements for manufacturers and Notified Bodies
Stricter requirements for manufacturers and Notified Bodies
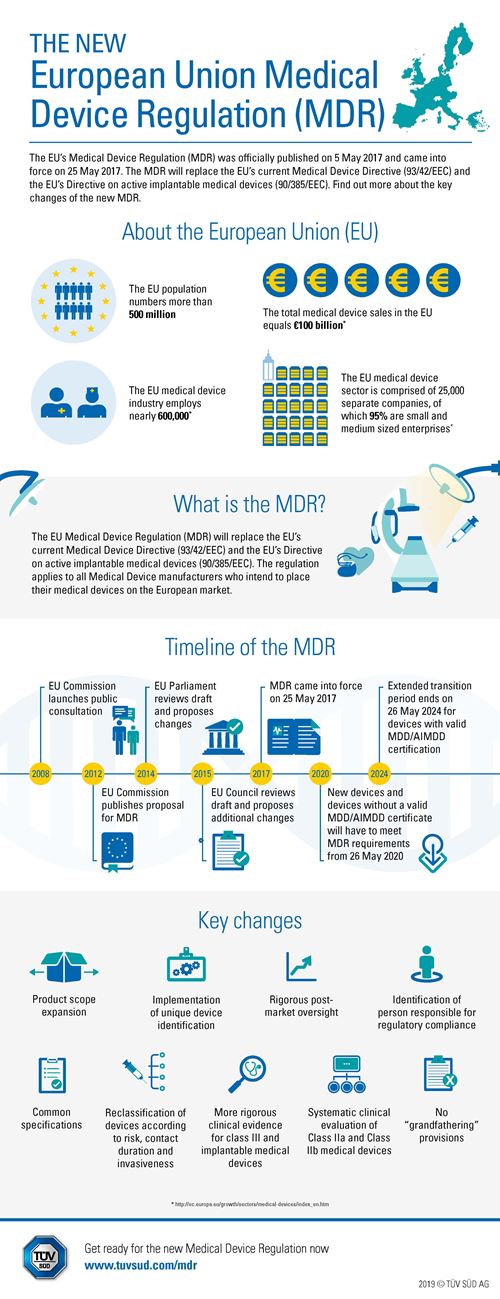
The new Medical Device Regulation (MDR, EU 2017/745) of the European Union replaces Directives 93/42/EEC on medical devices (MDD) and 90/385/EEC on active implantable medical devices (AIMD). “The regulation applies to all medical device manufacturers seeking to place their products on the EU market. It imposes strict demands on medical device manufacturers and the Notified Bodies which they must involve in the conformity assessment of medical devices other than self-declaration class I devices. We are proud that TÜV SÜD fulfils the strict requirements of the MDR and is now among the world's first Notified Bodies authorised to provide certification services under the new regulation”, says Dr Royth von Hahn, Global Head of Medical & Health Services (MHS) at TÜV SÜD Product Service.
The number of Notified Bodies currently under the ongoing designation process is fewer than the number of Notified Bodies designated under the MDD. Only 39 out of the current 58 Notified Bodies have applied for a new designation under the MDR. Of these, 23 Notified Bodies have been audited, and TÜV SÜD is now the second Notified Body to be designated.
Key changes contained in the MDR
In contrast to EU Directives, EU Regulations apply directly without having to be transposed into national law first. This significantly reduces the risk of different interpretations in individual EU Members States. The intention behind the new MDR is to improve the quality and safety of medical devices on the EU market. With this objective in mind, the requirements to be fulfilled by all stakeholders and especially medical device manufacturers were further specified and made stricter.
Overview: the main changes for manufacturers in the MDR
More precise and stricter requirements:
Regarding the content and scope of the technical documentation of medical devices and the manufacturers' quality management systems.
For clinical evaluations and clinical investigations, particularly regarding the type, scope and quality of clinical data.
Regarding post-market surveillance to be continuously included in product assessment.
More obligations regarding documentation and reporting to authorities and Notified Bodies (post-market surveillance plan/report, post-market clinical follow-up report, periodic safety update report, summary of safety and clinical performance).
Requirements concerning the clear registration, identification and traceability of medical devices (unique device identification, UDI).
The new MDR does not include a grandfathering clause. Products that are already on the market also need to meet the new requirements and pass the same conformity assessment process as new products. “Medical device manufacturers are well advised to inform themselves quickly about the requirements and deadlines of the new MDR. TÜV SÜD recommends that manufacturers of devices that have already been approved should contact their Notified Bodies in order to plan their conformity assessment according to the new requirements. This is a crucial step since the number of Notified Bodies able to deliver such services will be limited in future. Comprehensive preparation and early measures are key to ensuring a smooth transition to the new regulations”, emphasises Dr Bassil Akra, Vice President, Global Strategic Business Development of Medical & Health Services (MHS) at TÜV SÜD Product Service.
The role of the Notified Bodies according to the EU Regulation
As a fundamental rule, responsibility for ensuring that medical devices comply with legal requirements rests with the device manufacturers. For medical devices in higher risk classes, manufacturers need to involve a Notified Body in the approval process. The Notified Bodies must be designated by a national supervisory authority and notified by the EU Commission. The independence of the Notified Bodies is ensured by the notification system, which provides for continuous surveillance and regular re-designation by the supervisory authorities.
TÜV SÜD's international expertise
TÜV SÜD is one of the world's leading Notified Bodies for the approval of medical devices. Its more than 750 medical device professionals can be found at more than 30 locations throughout the world. Manufacturers benefit from both TÜV SÜD's technical, scientific and clinical expertise and its extensive international accreditations, including NRTL, INMETRO and the Medical Device Single Audit Program (MDSAP). These benefits considerably reduce the efforts involved in accessing international markets and time to market. The Medical Device Single Audit Program (MDSAP) is a project of the International Medical Device Regulators Forum (IMDRF). The programme enables manufacturers to ensure compliance with the regulatory requirements in several countries, i.e. Australia, Brazil, Japan, Canada and the USA, through a single quality-management audit.
Further information about the EU Medical Device Regulation, the key changes it involves and TÜV SÜD's services in this context can be found at https://www.tuvsud.com/mdr.
Press-contact: Dirk Moser-Delarami
Site Selector
Global
Americas
Asia
Europe
Middle East and Africa

