
如何实现医疗器械的欧盟市场准入
如何实现医疗器械的欧盟市场准入
TÜV SÜD has developed an online service registration form to allow us to systematically process your request. If you would like to request MDR services from TÜV SÜD, please use this form to register your interest.
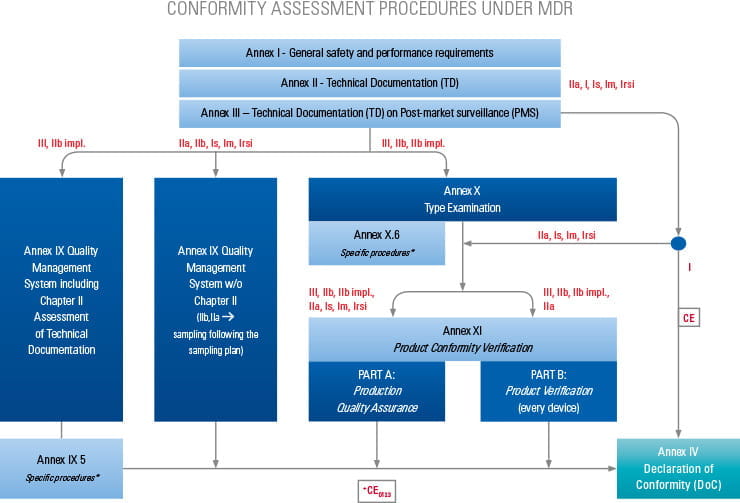
Based on the product classification, the manufacturer must apply for an applicable conformity assessment procedure.
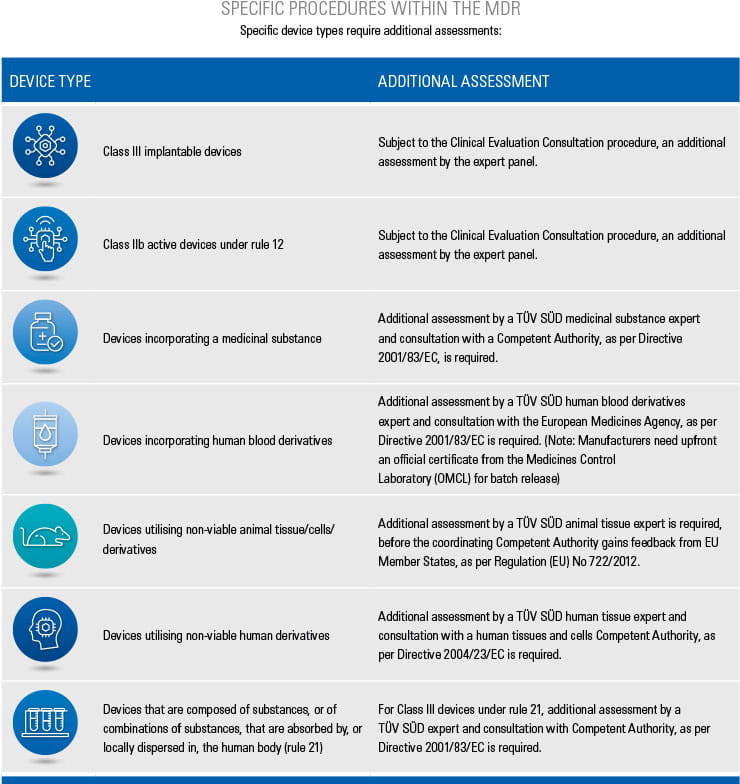
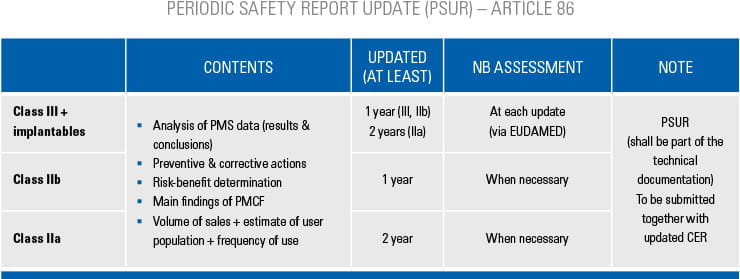
Step-by-step information for each of the conformity assessment procedures (using the relevant Annex) is highlighted below. The graphics also provide an overview of the procedures for different device classes and types as well as relevant surveillance activities. Specific device types require additional assessments, which are listed in the tables below.
English and/or German are the only acceptable languages for the submission of documentation and any related correspondence.
The certification costs are based on hourly rates and take into account factors such as the size of company, sites, number and complexity of devices, etc.
 *Class III implantable devices, class III with incorporated medicinal product, class III utilising tissues or cells of human or animal origin, class IIb active devices intended to administer and/or remove a medicinal product
*Class III implantable devices, class III with incorporated medicinal product, class III utilising tissues or cells of human or animal origin, class IIb active devices intended to administer and/or remove a medicinal product
+Refer to the Nando website for the applicable products and procedures/annexes.
CONFORMITY ASSESSMENT BASED ON A QUALITY MANAGEMENT SYSTEM AND ON THE ASSESSMENT OF TECHNICAL DOCUMENTATION
Chapter I: Quality Management System (QMS)
1. Application Management
2. Technical Documentation
(Assessment of the technical documentation is performed before or during routine audits)
Chapter II: Assessment of Technical Documentation (additional for classes III, IIb impl.)
2. Technical Documentation Assessment
3. Certification
2. Assessment of Technical Documentation
3. Testing
4. Certification
5. Additionally required: conformity assessment according to Annex XI-A XI-B
CONFORMITY ASSESSMENT BASED ON PRODUCT CONFORMITY VERIFICATION
1. Annex XI (Conformity assessment based on product conformity verification)
2. Annex XI in addition to Annex X (Conformity assessment based on product conformity verification and type-examination)
Part A Production Quality Assurance
1. Application Management
2. Auditing (Production QMS)
3. Technical Documentation (not applicable in cases where Annex XI is in addition to Annex X as this has already been assessed)
Please note: Assessment of the technical documentation is performed before or during the routine audits
4. Certification
Part B Product Verification
1. Application Management
2. Technical Documentation
4. Certification
Auditing clinical processes is an integral part of our QMS audits for all medical device manufacturers. The MDR has put even more emphasis on requirements related to clinical aspects. Consequently, focussed clinical audits are conducted as a risk-based surveillance activity within the regular conformity assessment process under the MDR. These clinical audits are targeted at medical device manufacturers of high-risk products (implants, class III and class IIb rule 12 devices) at least once every 3 years. In addition, and irrespective of the medical device class, clinical audits can be triggered in response to information that raises concern about the compliance and/or effectiveness of clinical processes of a medical device manufacturer. Clinical audits are performed by auditors, with specialised qualification and training in the field of clinical affairs.


On May 5th 2017, the European commission has published a new regulation for medical devices.
Learn more



Site Selector
Global
Americas
Asia
Europe
Middle East and Africa

